Research Group Bachmann-Gagescu
Genetics and Molecular pathogenesis of Ciliopathies
Our research focuses on a group of human disorders called ciliopathies, which are caused by the dysfunction of primary cilia, small non-motile organelles present on most vertebrate cells. Primary cilia serve as antennae transmitting sensory, mechanical or chemical information to the cell, and regulating signalling pathways during development and cell homeostasis.
Patients suffering from ciliopathies have a variety of clinical presentations, including symptoms such as retinal degeneration, cystic kidney disease, skeletal abnormalities, brain malformations and/or cognitive dysfunction. Hallmarks of ciliopathies are their prominent genetic heterogeneity, where each individual disorder can be caused by mutations in tens of genes, and their marked phenotypic variability, where individuals sharing the same causal mutations can display very different clinical pictures. These poor genotype-phenotype correlations significantly complicate the clinical management for individuals with ciliopathies, especially for predicting the outcome in the prenatal setting and for progressive features such as retinal degeneration.
The goal of our research is therefore to provide a better understanding of the molecular consequences of genetic variation in cilia-related genes. To reach this goal, we use a combined approach based on human genetics, iPSC-based cellular models and zebrafish genetics and biology.
Human genetics: The focus here is on Joubert syndrome, a ciliopathy characterized by a distinctive hindbrain malformation (“the molar tooth sign”) accompanied by intellectual disability and ataxia, as well as variable retinal dystrophy, fibrocystic kidney and liver disease and skeletal abnormalities.
Joubert syndrome is inherited in a recessive manner and can be caused by bi-allelic mutations in one of over 35 genes, many of which also cause other ciliopathies such as Meckel syndrome, nephronopthisis or Bardet-Biedl syndrome. This part of our research uses classical and next-generation human genetics techniques and phenotypic observation of patients with Joubert syndrome to try to recognize associations between genetic variation types and clinical outcome.
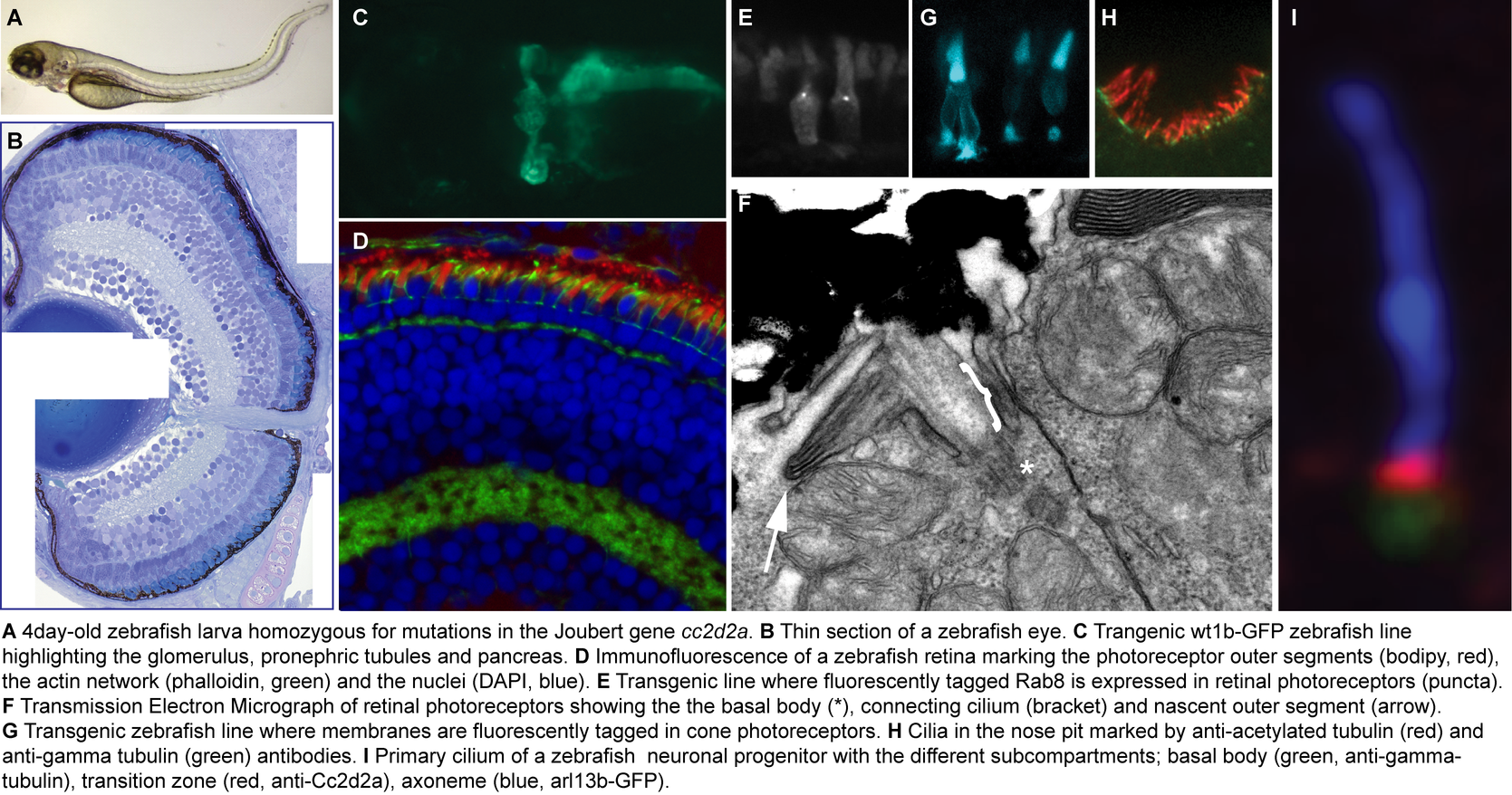
Animal model: We use zebrafish mutants in Joubert-associated genes to understand the function of the encoded proteins. A specific emphasis is placed on the retinal and renal phenotypes of zebrafish mutants given the excellent conservation of these organ systems across species.
The zebrafish has emerged as a major model for human development and disease thanks to its rapid external development and its accessibility to genetic analysis and manipulation. Phenotypic analysis is facilitated by transparency of the embryos providing excellent imaging conditions for direct observation of developmental processes under the microscope, and robust transgenesis techniques allowing in vivo examination of fluorescent markers tagging ciliary proteins of interest.
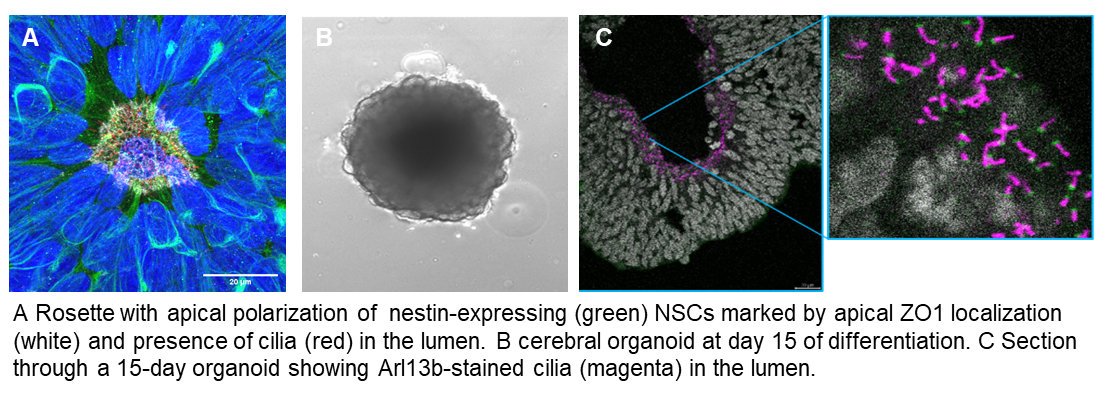
iPSC-based models: We are applying CRISPR-gene editing to iPSC lines and differentiate these towards the neuronal lineage following 2D and 3D protocols in order to investigate the function of Joubert-associated genes in human neuronal cells.
For more details, see iPSC-Research